From Category Theory to Enzyme Design:
Unleashing the Potential of Computational Systems Chemistry
This webpage provides an introduction to the project “From Category Theory to Enzyme Design: Unleashing the Potential of Computational Systems Chemistry”. The project is funded by the Novo Nordisk Foundation (2020-2022) as a grant under the Exploratory Interdisciplinary Synergy Programme. The project combines the expertise from the Algorithmic Cheminformatics Group at IMADA (Daniel Merkle, Rolf Fagerberg, Jakob L. Andersen), from the Fontana Lab at Harvard Medical School (Walter Fontana), and from the Department of Theoretical Chemistry (Christoph Flamm).
| Project Title: | From Category Theory to Enzyme Design: Unleashing the Potential of Computational Systems Chemistry |
| Applicant: | Prof. Daniel Merkle |
| Amount: | 4.997.700 DKK |
| Period: | 07/2020 - 12/2023 |
| Grant Number: | NNF19OC0057834 |
The project
Many important questions and challenges in research, industry, and society involve large and complex networks of chemical reactions. Some examples are: understanding the regulation of metabolic networks in humans; planning and optimizing chemical synthesis in industry and research labs; modeling the fragmentation of molecular ions inside mass spectrometers; developing personalized medicine; probing hypotheses of the origins of life; and monitoring environmental pollution in air, water and soil [1,2]. In this interdisciplinary and international project we will combine existing approaches from concurrency theory (Harvard Medical School, HMS) and graph rewriting (University of Southern Denmark) in order to develop ground-breaking new computational methods for analyzing networks of chemical reactions and thereby pave the way to applying these methods in research and industry. Together with chemists (University of Vienna) we will explore the applicability based on a carefully chosen flagship project: enzyme design.
Classical approaches to synthesis typically focus on small sets of molecules and often consider one reaction at a time in a planned sequence. Systems chemistry is a new emerging field that addresses the need to study networks of reactions, i.e., systems in which many distinct types of molecules can participate in many distinct chemical transformations. In such networks, reactions occur autonomously and asynchronously-without central coordination-whenever reactants are available. Systems of this kind are ubiquitous and include metabolic networks within organisms, networks of chemical production and exchange between organisms on an ecological scale, atmospheric chemistry and geochemistry, industrial production networks, and one-pot approaches to chemical synthesis. Due to the size and combinatorial complexity of these systems, it is unfeasible or outright impossible to manually analyze their properties and explore their design space. The field is therefore in strong need of new computational approaches and formal analytical methods to assist in modeling and design.
Any modeling approach must choose some level of abstraction, which determines the type of physical detail to be represented explicitly. E.g., a highly detailed level of resolution is provided by quantum chemistry. While the methods are highly accurate, they are also computationally very intensive, which restricts their practical deployment to single reactions between molecules of moderate size. The analysis of several interconnected reactions, let alone of large chemical reaction networks (CRNs), is presently out of reach. It is also unclear whether the questions that arise in the context of large reaction networks necessitate such a high level of resolution. At the same time, many extant computational methods for systems chemistry and biology are formulated at the other abstraction extreme, in which the structure of molecules is represented either not at all (molecules being simply assigned proper names) or in a very rudimentary fashion that does not permit the tracking of individual atoms across a series of reactions.
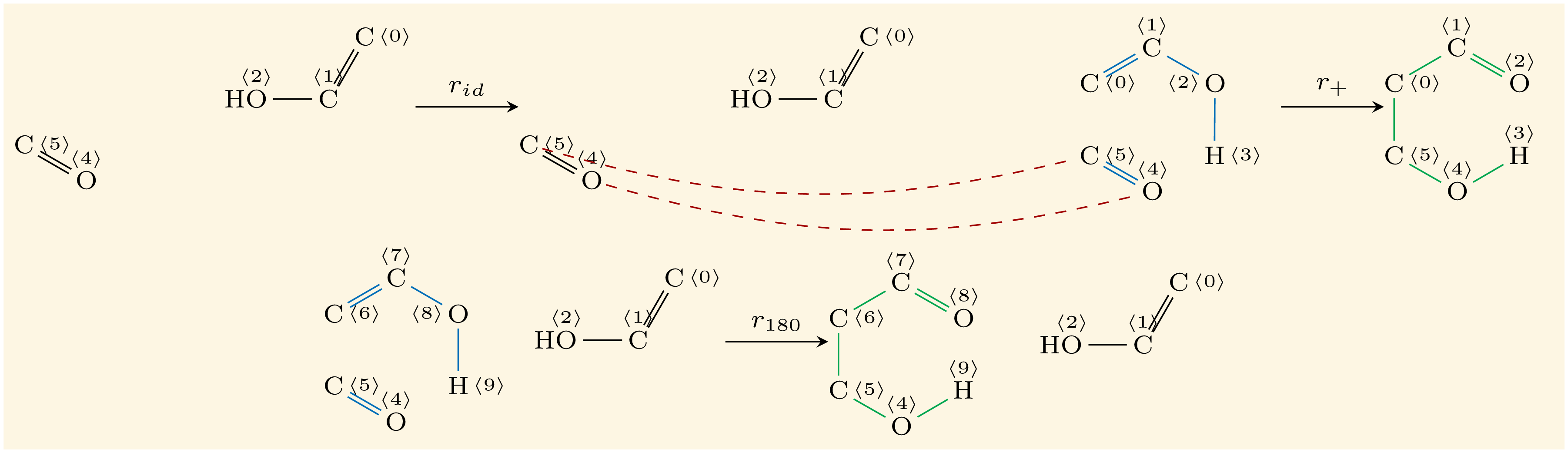
Example of application of a graph transformation rule: Double-Pushout (DPO) representation of a chemical reaction (Meisenheimer rearrangement). The top span $L \leftarrow K \rightarrow R$ defines a generic reaction, the bottom span is the application of the reaction to a set of educt molecules $G$ in order to infer a set of product molecules $H$. Here we chose an example with $|G|=|H|=1$, usually the educt and product set is larger. Note, that rule context inference $K$, i.e., the part that needs to exist but remains unmodified by applying a rule — in this example only the bond between oxygen and nitrogen — is defining how specific (but maybe over-specified)/generic (but maybe mechanistically wrong) a rule is.
The proposed project builds on a new and powerful methodology that strikes a balance between chemical detail and computational efficiency. The approach lies at the intersection of classical chemistry, present-day systems chemistry and biology, computer science, and category theory. It adapts techniques from the analysis of actual (mechanistic) causality in concurrency theory to the chemical and biological setting. Because of this blend of intellectual and technical influences, we name the approach computational systems chemistry (CSC). The term “computational” emphasizes both the deployment of computational tools in the service of practical applications and of theoretical concepts at the foundation of computation in support of reasoning and understanding. The goal of this exploratory project is to provide a proof-of-concept toward the long-term goal of tackling many significant questions in large and combinatorially complex CRNs that could not be addressed by other means. In particular, CSC shows promise for generating new technological ideas through theoretical rigor. This exploratory project is to be considered as initial steps towards establishing this highly promising area through the following specific objectives:
-
Integrate and unify algorithmic ideas and best practices from two existing platforms. One platform was conceived, designed, and implemented for organic chemistry by the lead PI and his group in Denmark as well as the chemistry partner from University of Vienna. The other platform draws on the theory of concurrency and was designed and implemented for protein- protein interaction networks supporting cellular signaling and decision-making processes by the partner from Harvard Medical School and his collaborators. The combination is ripe with potential synergies as both platforms are formally rooted in category theory.
-
Demonstrate a proof-of-concept (PoC) using a biochemical driving project. The goal of this exploratory project is the analysis and design of enzymes whose catalytic site is viewed as a small (catalytic) reaction network in its own right. Such enzymes can then be used in the design of reaction networks.
-
Train the next generation of scientists for CSC: This will enable the transition towards a large-scale implementation of our approaches to tackle key societal challenges, such as the development of personalized medicine, the monitoring of pollution, and the achievement of a more environmentally friendly and sustainable network of industrial synthesis.
We argue that CSC is in a position today similar to where bioinformatics and computational biology were a few decades ago and that it has similarly huge potential. The long-term vision is to unleash that potential.
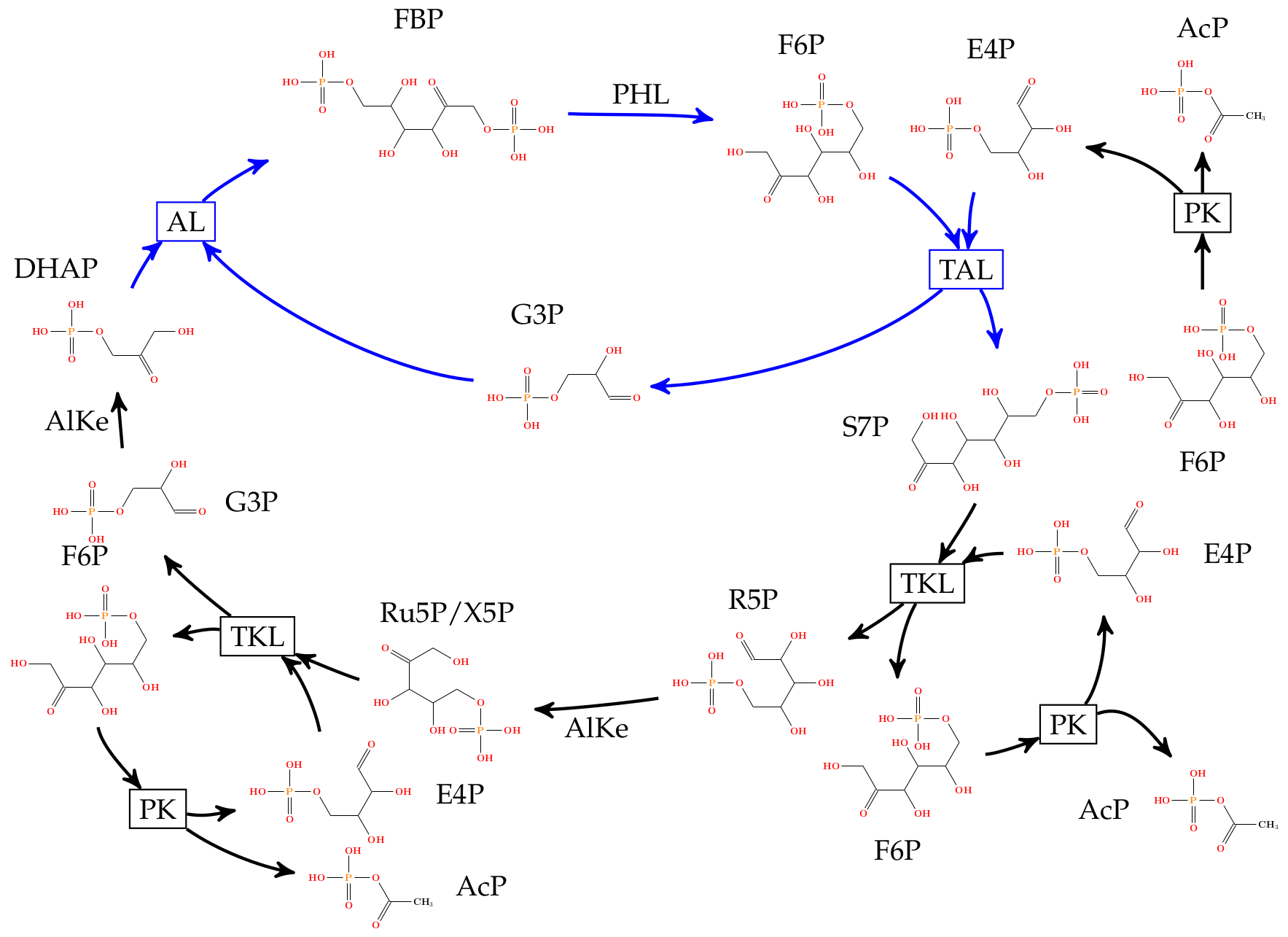
An automatically inferred pathway for a carbon-efficient efficient (no carbon loss) alternative to glycolysis. The solution is similar to the engineered solution suggested in (Bogorard et al., 2013), which was successfully implemented in vitro and in vivo. Note, that our approach allows to infer many more solutions (see [3]) with any additional mathematically defined constraints. Fructose-6-phosphate (F6P) and acetylphosphate (AcP) are input compounds, a large chemical space is expanded (graph grammar rules depicted as boxes indicating the reaction name), and a catalytic solution is automatically inferred.
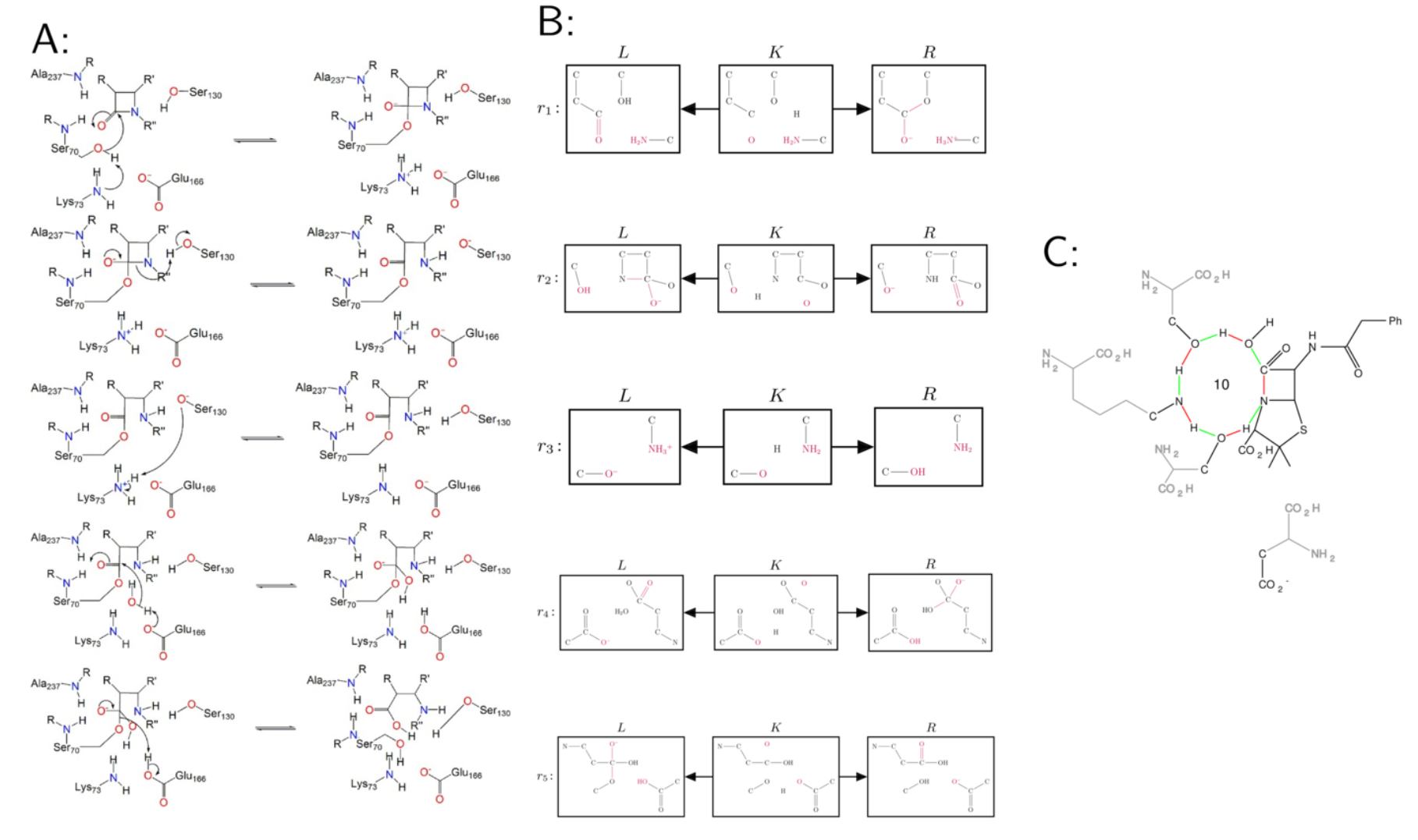
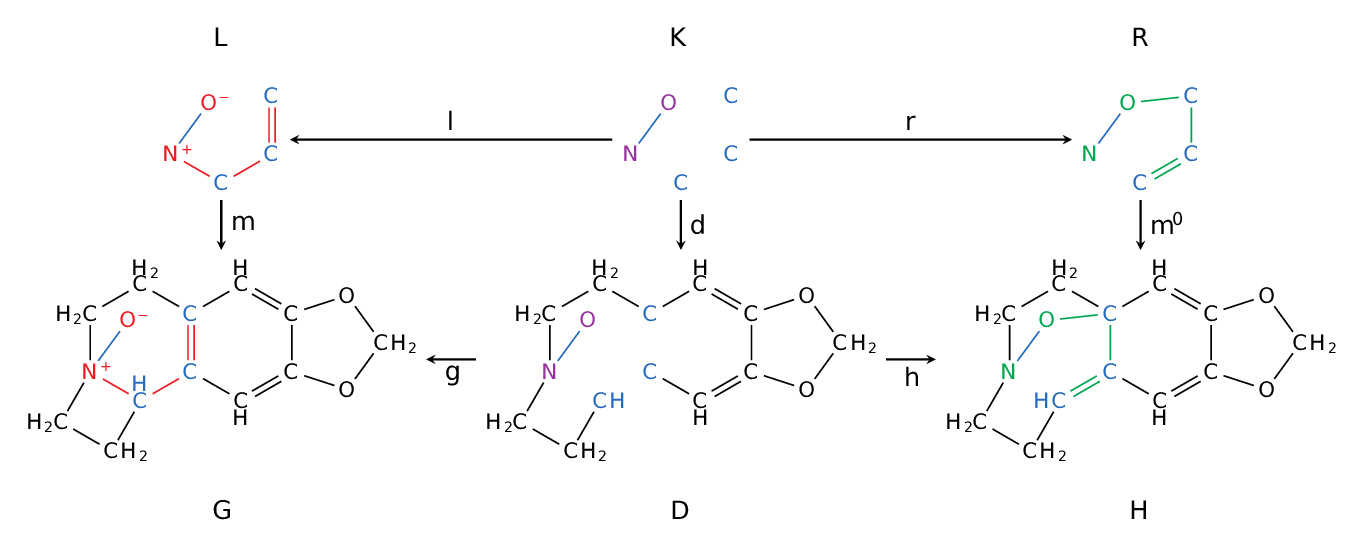
Five steps of $\beta$-Lactamase. A: electron pushing diagrams from the M-CSA databse. B: manually created DPO rules. C: an example of an overall reactions resulting from composing the graph rewrite rules for the elementary steps of the $\beta$-lactamase (formally $r=i_G \circ r_1 \circ r_2 \circ r_3 \circ r_4 \circ r_5\circ i_H$), red edges indicate bonds broken, green edges infer bonds formed), see [4].
References
-
Computational Chemistry: The Fate of Current Methods and Future Challenges
S. Grimme and P.R. Schreiner
Angew. Chem. Int. Ed., 57.16 (2017), 4170–76 -
Rewiring Chemistry: Algorithmic Discovery and Experimental Validation of One-Pot Reactions in the Network of Organic Chemistry
Gothard C.M. et al.
Angewandte Chemie, 51(32), 7922–27, 2012 -
Chemical Transformation Motifs --- Modelling Pathways as Integer Hyperflows
Andersen, J. L., C. Flamm, D. Merkle and P. F. Stadler
IEEE/ACM Trans. Comput. Biol. Bioinform., 16(2):510-523, 2019 -
Rule Composition in Graph Transformation Models of Chemical Reactions
Andersen, J. L., C. Flamm, D. Merkle and P. F. Stadler
MATCH Communications in Mathematical and in Computer Chemistry, 80.3 (2018), 661–704
Publications
-
Atom Tracking Using Cayley Graphs
Marc Hellmuth, Daniel Merkle, Nikolai Nøjgaard
Proceedings of Bioinformatics Research and Applications : 16th International Symposium, ISBRA 2020, 2020 [ DOI, TR ]
-
Cayley Graphs of Semigroups Applied to Atom Tracking in Chemistry
Nikolai Nøjgaard, Walter Fontana, Marc Hellmuth, Daniel Merkle
JournalJournal of Computational Biology, 28(7), 2021 [ DOI, TR ] -
Graph transformation for enzymatic mechanisms
Jakob L. Andersen, Rolf Fagerberg, Christoph Flamm, Walter Fontana, Juraj Kolčák, Christophe V. F. P. Laurent, Daniel Merkle, and Nikolai Nøjgaard
Bioinformatics, 37(Supplement_1), 2021 [ DOI, TR ]
-
Representing Catalytic Mechanisms with Rule Composition
Jakob L. Andersen, Rolf Fagerberg, Christoph Flamm, Walter Fontana, Juri Kolčák, Christophe V. F. P. Laurent, Daniel Merkle, and Nikolai Nøjgaard
Journal of Chemical Information and Modeling, 62(22), 5513-5524, 2022 [ DOI, TR ]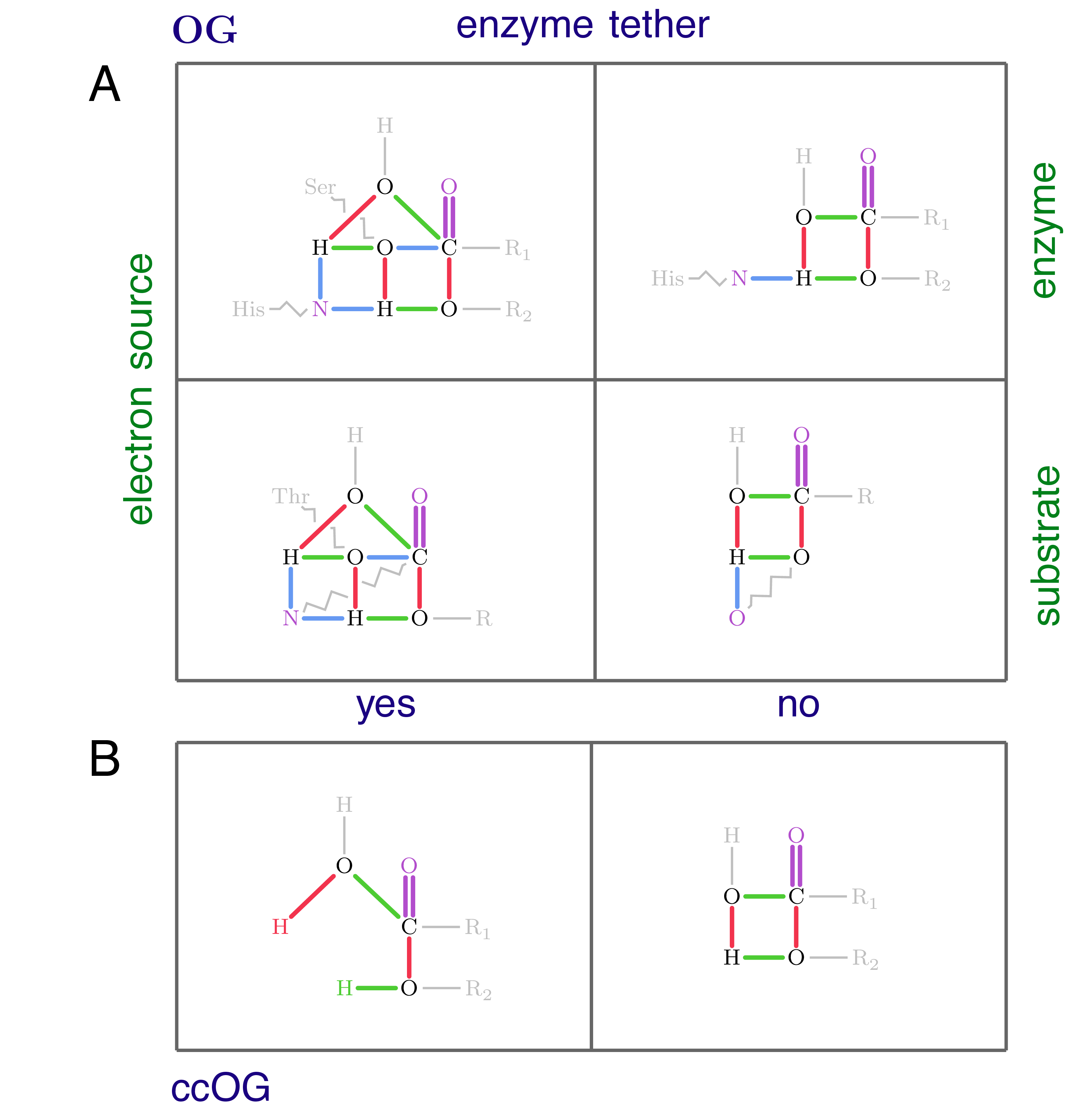
-
Rewriting theory for the life Sciences: A unifying theory of CTMC semantics
Nicolas Behr, Jean Krivine, Jakob L. Andersen, Daniel Merkle
Theoretical Computer Science, 884, 68-115, 2021 [ DOI, TR ]